レメロン®臨床成績 長期投与試験
レメロン®臨床成績 長期投与試験
「禁忌を含む使用上の注意」等についてはDIをご参照ください。
臨床成績:レメロン®国内第Ⅱ相長期投与試験(オープン試験)
(1)試験概要
長期投与試験(承認時評価資料)
| 目 的: | うつ病及びうつ状態の患者を対象に、レメロン®の長期投与による安全性(有害事象の発現率)及び有効性を検討する。 |
| 対 象: | プラセボ対照比較試験に参加した患者のうち投与終了時に臨床全般印象度(CGI)が「やや良くなった」以上と評価されたうつ病及びうつ状態の患者109例 |
| デザイン: | 多施設共同オープンラベル試験 |
| 投与方法: | レメロン®は投与開始から1週間は15mg/日とし、2週目以降は忍容性に問題がないことを確認した上で増量可能とした。1回の増量範囲は15mgとした。なお、投与量は症状に応じ1日15~45mgの幅で適宜増減できるものとした。投与期間は52週間とし、1日1回、就寝前に経口投与することとした。 |
| 評価項目: | 有効性評価項目:投与終了(中止)時におけるHAM-D合計スコア(17項目) 安全性評価項目:有害事象及び副作用の発現状況 |
| 解析計画: | 109例中、HAM-D合計スコア(17項目)において投与開始時かつ投与後最低1回が評価可能な107例が、有効性の最大解析対象集団(FAS)となった。 |
| 判定基準: | HAM-D合計スコア(17項目)の変化 |
(2)有効性
HAM-D合計スコア(17項目)を用いて、投与開始前、各観察時及び投与終了(中止)時に評価を行い、このうち投与終了(中止)時におけるHAM-D合計スコア(17項目)を有効性評価項目とした。HAM-D合計スコア(17項目)は、投与開始前の10.2±6.5(n=107)から投与開始6週では、6.9±5.1となった(OC:ObservedCases、観察時データ)。投与6週後以降、投与52週後(4.0±5.2)まで7未満で推移した。
■HAM-D合計スコア(17項目)(OC)
| 観察時点 | 投与開始前 | 投与6週後 | 投与12週後 | 投与24週後 | 投与52週後 |
| 症例数 | 107 | 100 | 97 | 83 | 71 |
| HAM-D 合計スコア注1) | 10.2 (6.5) | 6.9 (5.1) | 5.7 (4.9) | 4.1 (4.1) | 4.0 (5.2) |
注1):平均値(標準偏差)
■各観察時点のHAM-D合計スコア(17項目)の推移(OC)
の推移(OC)")
(3)安全性
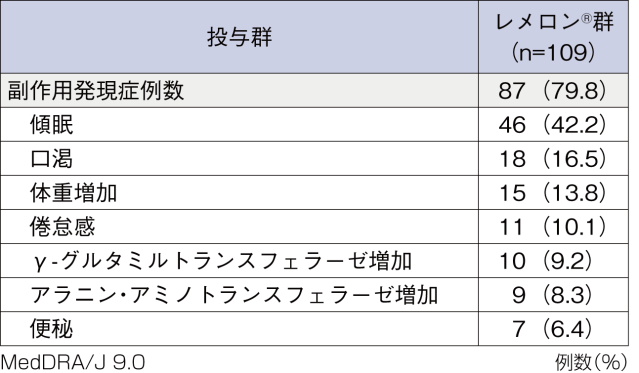
安全性評価対象症例109例中87例(79.8%)に副作用が認められた。主なものは、傾眠46例(42.2%)、口渇18例(16.5%)であった。
死亡に至った副作用は認められなかった。重篤な副作用は、アラニン・アミノトランスフェラーゼ増加及びアスパラギン酸アミノトランスフェラーゼ増加1例、交通事故1例であった。投与中止に至った副作用は4例に6件(傾眠、倦怠感、パニック発作、血中ビリルビン増加、γ-グルタミルトランスフェラーゼ増加及び軽躁が各1件)が認められた。
■5%以上発現した副作用